
Dans le cadre de cette enquête, le Consortium a examiné les données de millions de signalements rapportés par les fabricant·es et autres, principalement par la Food and Drugs Administration (FDA). Lors de la dernière décennie, près de 500.000 alertes en lien avec un dispositif médical.ont été enregistrées.
La base de données suivante regroupe les produits susceptibles d’être commercialisés en Tunisie* et sur lesquels une action a été requise (mise en garde auprès des sociétés distributrices, rappels de certains lots, actions correctives, etc.).
Template Not found:
@inkube/component/null
Sur le même sujet

Les retraits de produits
Que ce soit du côté des fournisseur·es, des institutions nationales de contrôle ou des praticien·nes, tous et toutes l’affirment : en Tunisie les retraits des dispositifs médicaux non conformes sont systématiques et effectués dans les règles. Pourtant, malgré une procédure claire, les patient·es concerné·es sont les dernier·es informé·es des risques que peuvent représenter certains produits.
La majorité des dispositifs médicaux distribués en Tunisie sont fabriqués à l’étranger et importés. Lorsqu’un produit rencontre un problème à travers le monde, le ou la fabricant·e doit informer les pays concernés à travers ses distributeurs.
Zinelabidine Mahsni, PDG de Toucomex, société distributrice de Medtronic, explique qu’en fonction du problème, les actions peuvent être de trois types : préventive, corrective ou le retrait du produit.
Dans le premier cas, le fabricant demande aux praticien·nes d’être vigilant·es car le dispositif présente des problèmes d’utilisation dont la cause n’a pas été identifiée. Si la cause est connue, il faut appliquer une “action corrective” et les praticien·nes sont informé·es des manipulations à effectuer pour éviter le problème. Enfin, en cas de retrait, certains lots ou le produit dans son ensemble doivent être retirés de la commercialisation.
Les patient·es ne sont éventuellement informé·es qu’à travers leur médecin, si il ou elle juge l’information nécessaire.
Un implant cardiaque distribué en Tunisie et fabriqué par Medtronic a par exemple fait l’objet d’un action corrective pour avoir fait 19 morts et 39 blessés à travers le monde. Un chiffre que le PDG de Toucomex nuance : “Cela ne représente que 0,136% cas” sur l’ensemble des produits distribués.
À partir de 0,1% d’incidents, Medtronic entreprend généralement une action corrective mais n’est pas obligé de retirer son produit du marché : les praticien·nes doivent simplement “corriger leur utilisation” du dispositif et veiller à son bon fonctionnement. Ce seuil de risque se situe généralement entre 0,1% et 0,2% selon les entreprises.
Lorsqu’une alerte de ce type est lancée par le ou la fabricant·e, les distributeurs et distributrices doivent en informer l’agence nationale de contrôle sanitaire et environnemental des produits (ANCSEP). En collaboration avec la Direction de la pharmacie et du médicament (DPM), ces deux entités sont chargées de centraliser les informations et vérifier que les praticien·nes sont bien mis·es au courant des procédures à suivre.
Mais rien n’assure que le ou la patient·e soit bien au fait des risques encourus dans le cas où il ou elle serait concerné·e par ce dispositif. Pour une responsable du groupe KT Bio, société distributrice de Boston Scientific Corporation, il vaut mieux ne pas communiquer ce nombre d’incidents “pour ne pas affoler les patients”.
Pour Youssef Makni, président du conseil national de l’ordre des médecins, le ou la patient·e doit être mis·e au courant “en fonction du risque”.
“C’est la problématique du ‘consentement éclairé’, qui fait couler beaucoup d’encre à travers le monde”, détaille le médecin, “Aux États-Unis, on considère que le patient doit tout savoir en détails. Dans les pays européens et en Tunisie, on estime que le patient doit être informé des risques s’ils sont vraiment importants. Le but est d'éviter de l’affoler”.
“Le patient doit être protégé mais la gestion des risques relève de la responsabilité directe du praticien”, estime aussi Nadia Fenina, directrice de la DPM.
La DPM a pour rôle de publier une liste de produits non conformes sur son site internet. Entre 2012 et 2018, cette liste indique qu’une cinquantaine de dispositifs ont été retirés du marché tunisien, selon les données de septembre 2018. Pour la plupart, ils sont issus de laboratoires nationaux. C’est le laboratoire ADHE-ELS qui est le plus concerné par ces retraits avec une vingtaine de produits retirés entre 2012 et 2018.
Mais les produits indiqués concernent uniquement les dispositifs à usage unique : on retrouve dans la liste plusieurs types de sondes, des cathéters, des prothèses ou encore des compresses chirurgicales. D’autres dispositifs comme les scanners sont gérés par la Direction des équipements et ne sont pas indiqués sur cette base de données.
De plus, cette liste ne concerne que les retraits définitifs. Si le dispositif a simplement nécessité une action préventive ou corrective, il n’y sera pas mentionné même s’il a pu provoquer des morts et des blessé·es.
Une matériovigilance limitée
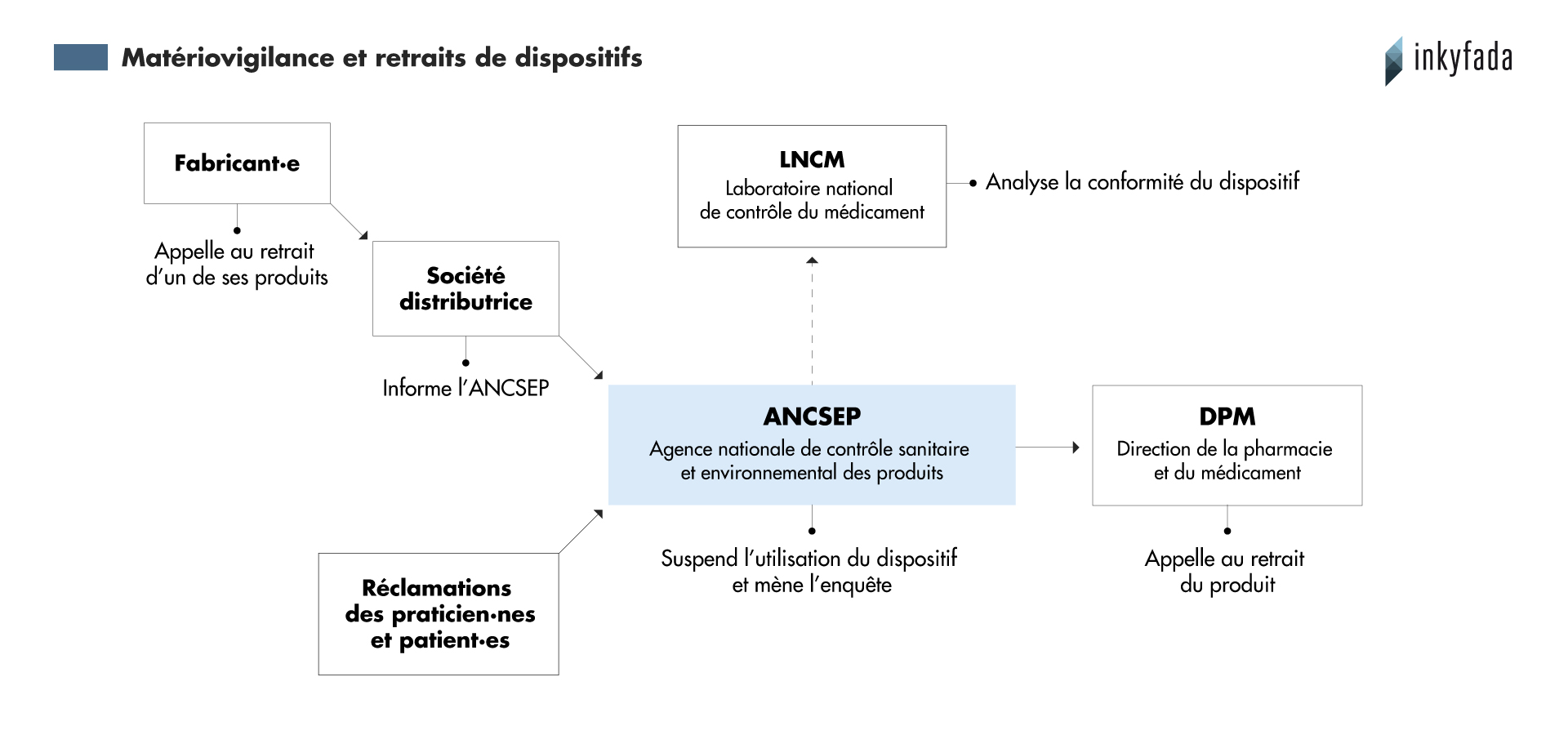
En plus des alertes initiées par les fabricant·es et agences étrangères, en Tunisie, l’agence nationale de contrôle sanitaire et environnemental des produits (ANCSEP) a pour rôle de recevoir les “réclamations faites par les médecins, les pharmaciens voire les patients”, explique Bochra Bejaoui, responsable à l’ANCSEP. L’agence assure aussi une veille des rappels publiés par la FDA et vérifie si les produits signalés concernent la Tunisie.
En moyenne, l’ANCSEP traite 75 déclarations par an. Entre 2011 et 2017, 325 incidents ont été traités par l’agence.
“Ce n’est pas suffisant, il y a un phénomène de sous-déclaration”, considère la responsable.
Selon elle, cela est dû au délai nécessaire pour fournir les résultats. “On met six à huit mois pour répondre”, car les analyses effectuées par le Laboratoire national du contrôle du médicament (LNCM) mettent beaucoup de temps à être effectuées.
En 2013, une étude de l’ANCSEP a montré que les produits les plus concernés sont principalement les cathéters mais aussi les bandes adhésives et les sondes.
Dès qu’un produit est signalé, l’agence doit contacter la société distributrice. “On lui demande de nous fournir la liste des établissements ayant reçu le lot ou le même lot de dispositif incriminé”, continue-t-elle, “et on enquête pour déterminer s’il s’agit d’un mésusage ou d’une non-conformité”.
À travers la DPM et les fournisseur·es, une alerte est lancée à l'ensemble des professionnel·les concerné·es, leur demandant d’arrêter d’utiliser le dispositif jusqu’à la clôture de l’enquête.
L’ANCSEP détermine en interne si le problème provient d’une erreur humaine ou du produit en lui-même. Si c’est un problème de conformité, un échantillon est envoyé au LNCM tandis que le reste du lot incriminé reste en quarantaine. Enfin, si le laboratoire conclut qu’il est nécessaire de retirer le produit du marché, la DPM appelle à son retrait.
Une législation floue
En Tunisie, il n’existe pas de définition ni de législation claires en ce qui concerne les dispositifs médicaux. Bien qu’ils soient en apparence soumis à un contrôle strict, il n’existe pas de sanction d’un point de vue juridique en cas de défaillance d’un produit.
Les dispositifs médicaux constituent l’ensemble des équipements médicaux tels que les implants, les prothèses ou encore les scanners. “À part quelques produits de ligatures, tous les dispositifs médicaux sont importés”, explique Lotfi Ben Yedder, président de la chambre syndicale de l’équipement médical et hospitalier, “le marché local tunisien est très restreint”.
Ces produits sont généralement achetés après un appel d’offres. Les pharmacien·nes en milieu hospitalier expriment leurs besoins et les sociétés distributrices des grandes marques internationales font des propositions. “Dans le cas où le produit n’a pas d’équivalent sur le marché, la vente se fait de gré-à-gré”, explique Myriam Guerfali, pharmacienne hospitalière et présidente de l’association tunisienne des pharmaciens hospitaliers (ATPH).
Une fois la vente convenue, les dispositifs, fabriqués à l’étranger, sont importés puis vendus aux hôpitaux et cliniques privées. “On ne connaît pas le nombre de distributeurs en Tunisie car il n’y a pas vraiment de contrôle”, dit Lotfi Ben Yedder, “certaines entreprises ont des patentes ‘distribution de tout produit’ ou ‘import-export’”. Ces sociétés n’ont pas besoin d’être spécialisées dans la distribution de dispositifs médicaux pour en vendre.
En tant que président de la chambre syndicale de l’équipement médical et hospitalier, Lotfi Ben Yedder déplore ce manque de rigueur dans l’intitulé des activités. “On a longtemps milité pour que les distributeurs soient obligés de se spécialiser dans les dispositifs médicaux, mais sans résultat”, déplore-t-il, “ils peuvent importer ce qu’ils veulent. Mais heureusement, il y a un contrôle strict des produits à l’importation”.
Des produits bloqués pendant des mois
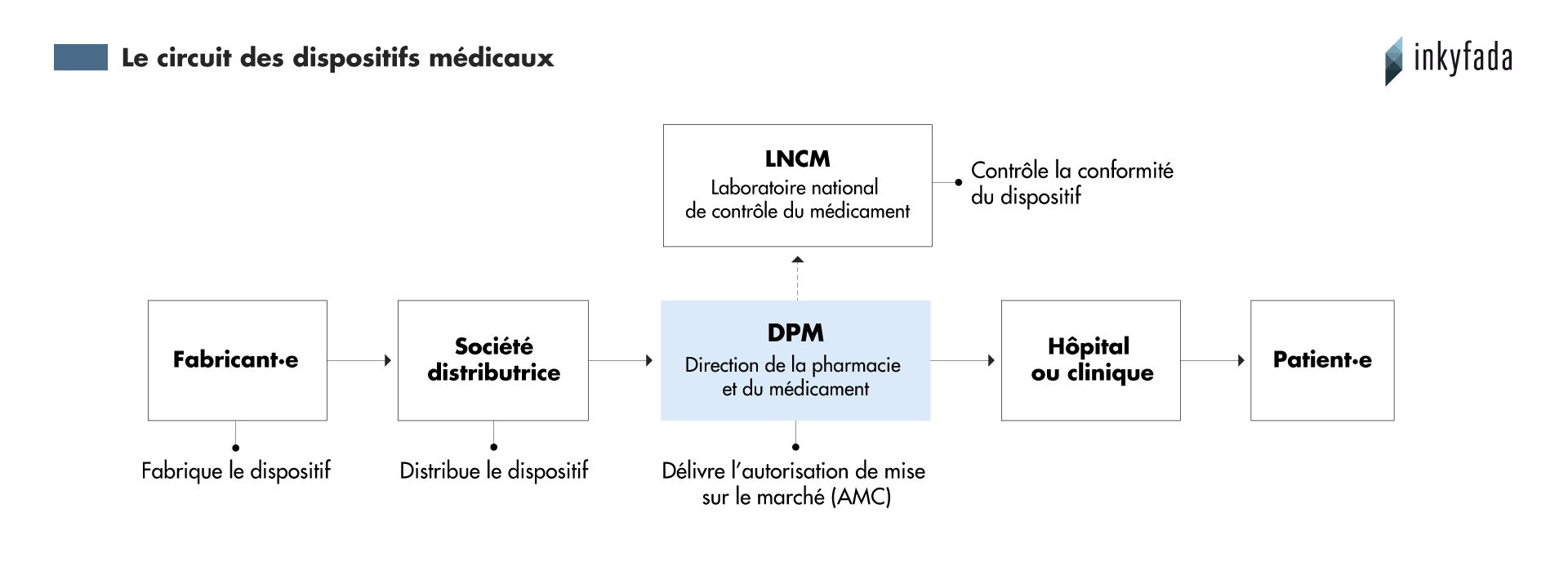
Ces produits n’arrivent pas aussi simplement sur le marché tunisien. Pour pouvoir vendre ces dispositifs, il est nécessaire d’obtenir une autorisation de mise à la consommation (AMC) délivrée par la Direction de la Pharmacie et du Médicament (DPM), une entité du ministère de la Santé. Cette institution travaille en collaboration avec le Laboratoire national de contrôle des médicaments (LNCM), chargé d’analyser la conformité de ces dispositifs.
Contrairement aux autorisations délivrées pour les médicaments, celle concernant les équipements médicaux n’est pas valable pour plusieurs années sur un produit donné : il est nécessaire de l’obtenir à chaque livraison de lot.
“Si le dispositif est importé régulièrement depuis plusieurs années, il doit juste recevoir un avis favorable du laboratoire”, explique Nadia Fenina, directrice de la DPM, “sinon il faut l’analyser, ce qui prend plus de temps”. Dans le premier cas, le ou la fabricant·e doit payer 35 dinars par lot. Si une analyse est nécessaire, cela coûte 200 dinars.
Cette systématisation du contrôle des dispositifs surcharge les services de la DPM et du LNCM. “Depuis avril 2014, tous les dispositifs implantables sont soumis à l’AMC”, détaille la pharmacienne Myriam Guerfali, “c’est une bonne chose, mais d’un coup, le LNCM s’est retrouvé avec beaucoup plus de dispositifs à vérifier !”
Cette autorisation met entre deux et quatre mois à être délivrée. En attendant, le produit est stocké plusieurs semaines en laboratoire sans pouvoir être utilisé. Ce délai affecte les fabricant·es, les praticien·nes mais surtout les patient·es.
“Vous imaginez quelqu’un qui attend un stent ? Il ne peut pas attendre quatre mois !”, s’exclame la pharmacienne.
Dans ces cas-là, les praticien·nes ont le droit de demander une priorisation de leur commande. “Parfois, la DPM peut donner une autorisation en moins de 24h si la demande est vraiment urgente”, explique Lotfi Ben Yedder, le président de la chambre syndicale, “mais on ne peut pas s’amuser à faire ça à chaque fois qu’il y a une urgence”.
En plus de payer les frais de l’AMC, les fabricant·es doivent régulièrement payer des pénalités de retard dans le cadre des appels d’offres.
Un contrôle retardé
En attendant l’obtention de l’AMC, les échantillons de dispositifs médicaux sont stockés au LNCM. Ils y restent plusieurs semaines, voire plusieurs en mois, en attendant de pouvoir être analysés. “Le problème, c’est que ces institutions de contrôle ne sont pas correctement outillées et manquent clairement de moyens”, justifie Lotfi Ben Yedder, “et la quantité de produits à contrôler est énorme, elles manquent aussi de capacités humaines”.
Pour désengorger ces services de contrôle, une commission du ministère de la Santé, composée de fonctionnaires, fabricant·es et praticien·nes, souhaite mettre en place un système de référencement des dispositifs médicaux jugés conformes. “Pour éviter d’être bloqués, on voudrait que les fournisseurs avec cinq AMC conformes sur le même produit soient référencés dans une base de données”, explique Myriam Guerfali. Si ces critères sont remplis, une AMC pourrait être immédiatement donnée pour certains produits.
Pour la pharmacienne, il est important de réduire au maximum le délai d’autorisation, quitte à réduire les contrôles. “On parle de marques internationales, vérifiées et re-vérifiées par la FDA et derrière, on re-contrôle chaque lot. Pourquoi ?” s’interroge-t-elle, “Et l’aberration, c’est que les produits locaux n’ont pas besoin de cette autorisation !”.
Les rares dispositifs fabriqués en Tunisie n’ont en effet pas besoin d’AMC : ils peuvent subir des inspections aléatoires ainsi que des contrôles post-marketing en cas de réclamation mais leur mise sur le marché n’a pas besoin d’être validée par la DPM et le LNCM. Une différence qui n’empêche pas que “les contrôles sur la fabrication locale soient très bien faits” selon Lotfi Ben Yedder.
Dans tout ce système de contrôle, les patient·es sont généralement laissé·es pour compte. L’accès à l’information n’est pas la priorité et l’évaluation des dangers reste du ressort exclusif des professionnel·les du secteur.
Sur plus de 15.000 produits compilés par la FDA dans leur base de données, au moins 900 produits seraient susceptibles d’être commercialisés en Tunisie et d’affecter les patient·es. Ces produits s’ajoutent aux dizaines de signalements enregistrés par l’ANCSEP ainsi qu’à la cinquantaine de retraits listés par la DPM depuis 2012.
Même si tout semble être mis en place pour assurer leur sécurité, il est difficile pour un·e citoyen·ne tunisien·ne d’obtenir des informations fiables et centralisées sur les incidents et les risques que représentent certains dispositifs médicaux.




